Research Overview
Project 1: KU70-Mediated Nucleus-Cytoplasm Communication in DNA Damage Response
Double-strand breaks (DSBs) are among the most damaging types of DNA damage induced by ionizing radiation (IR), profoundly disrupting the genome and serving as a key target for cancer therapies. In mammalian cells, DSBs are predominantly repaired via the non-homologous end joining (NHEJ) pathway, which takes precedence over homologous recombinational repair (HRR) to ensure cell survival following IR exposure. At the heart of this process is the Ku70 protein, known not only for its vital role in repairing DSBs but also for its unexpected functions in the cytoplasm, where it regulates apoptosis and orchestrates innate immune responses.
Building on our previous findings, we uncovered a novel mechanism: methylation of lysine 570 in the C-terminal nSAP domain of Ku70 triggers a dramatic translocation of the protein from the nucleus to the cytoplasm (Wang et al., Cell Reports, 2022). This translocation is more than a simple shift, it acts as a crucial survival signal, helping the cell resist apoptosis during genomic stress. We envision this methylation as a molecular messenger, guiding Ku70 to bolster cellular resilience in times of DNA damage. Interestingly, deleting the entire C-terminal SAP domain has little impact on normal development, yet it significantly increases cellular and organismal sensitivity to IR and chemotherapeutic agents that induce DSBs (Wang et al., NAR, 2025).
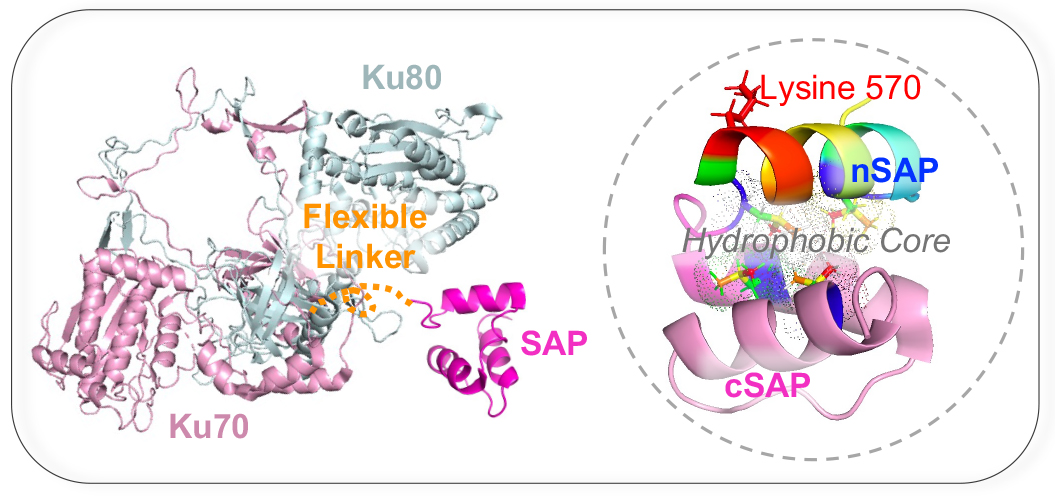
The structures of the human Ku70/Ku80 dimer (PDB ID: 7AXZ) and the Ku70 SAP domain (PDB ID: 1JJR) were from the Protein Data Bank. The models were modified and visualized using PyMoL.
Our central hypothesis is that methylation of lysine 570 in humans (or lysine 568 in mice), modulates the Ku70-SAP interaction, destabilizing the protein and impairing its associated NHEJ factors. This destabilization may promote Ku70’s migration into the cytoplasm, and disruption of this finely balanced process could result in inefficient DSB repair and compromised cytoplasmic functions. In Dr. Wang’s laboratory, we aim to explore the complex communication between the nucleus and cytoplasm and its critical role in the DNA damage response.
- Aim 1: To investigate how the absence of Ku70-nSAP methylation impacts susceptibility to apoptosis, inflammation, and defective DNA repair, using a newly generated Ku70 knock-in mouse model.
- Aim 2: To apply structural insights to develop strategies that destabilize SAP folding or weaken SAP-partner interactions within its hydrophobic core, thereby increasing cellular sensitivity to DNA damage.
The completion of this study will shed light on the elegant interplay between nuclear and cytoplasmic signaling mediated by Ku70 SAP domain or its methylation, opening new avenues for modulating Ku70 function and enhancing cancer therapies.
Project 2: Differential Radiation Responses of Bone Marrow Niche and Hematopoietic Cells Driven by SETD4 Enzymatic Function
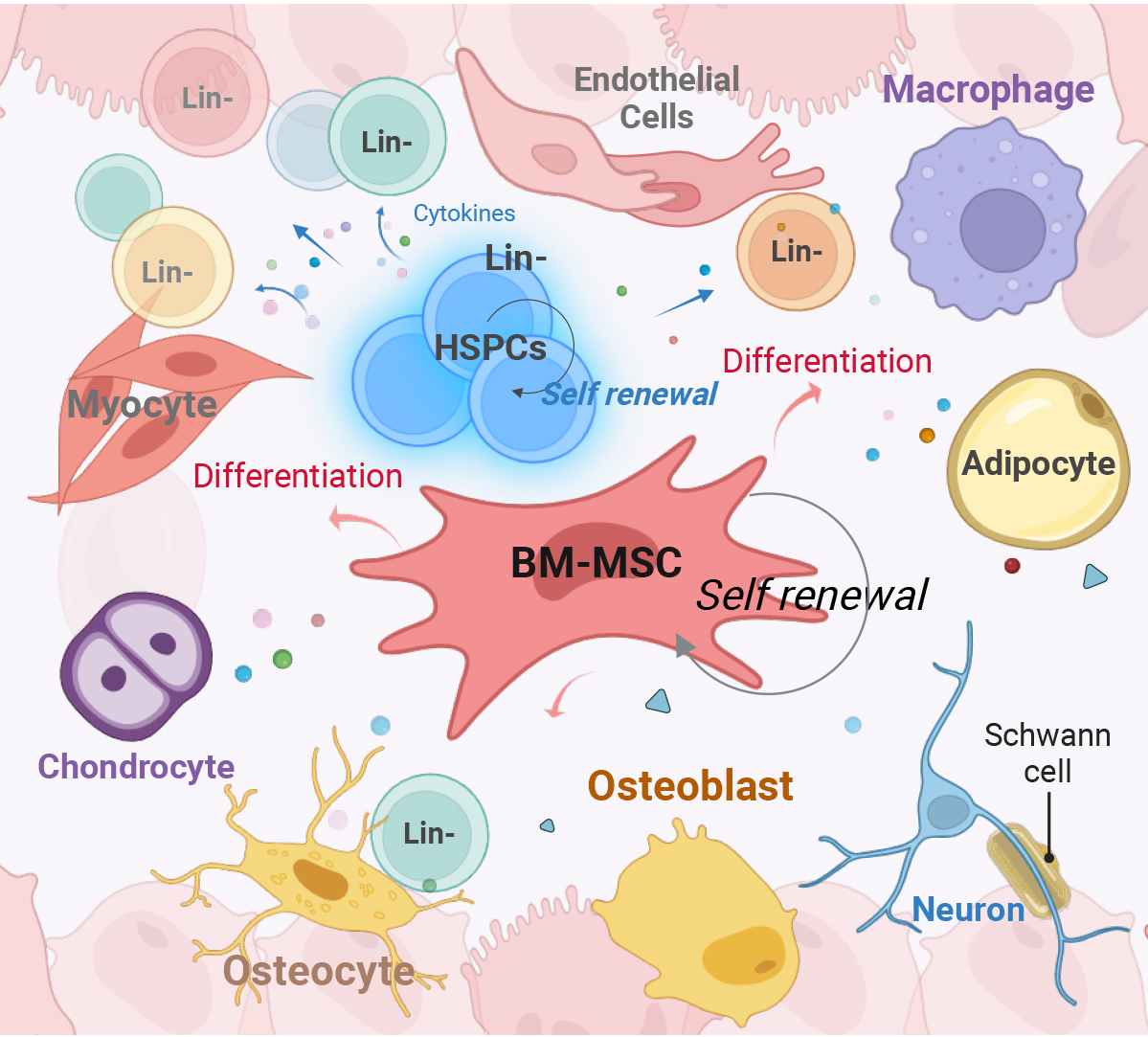
Schematic of the bone marrow (BM) niche environment surrounding Lineage-negative (Lin-) cells, including hematopoietic stem and progenitor cells (HSPCs). This intricate network of niche cells, featuring BM mesenchymal stem cells (MSCs) that differentiate into various cell types, collectively supports HSPCs and maintains hematopoiesis.
Patients with blood malignancies or hematopoietic disorders often require pre-conditioning regimens to eliminate diseased hematopoietic cells while preserving the bone marrow (BM) niche, ensuring successful engraftment and recovery. Total body irradiation (TBI) remains a cornerstone of pre-transplant conditioning, effectively eradicating malignant and dysfunctional hematopoietic stem/progenitor cells (HSPCs) in hematopoietic cell transplantation (HCT). The success of this approach depends on the differential sensitivity of recipient hematopoietic cells and the supportive BM microenvironment. Therefore, identifying molecular targets that can selectively modulate these sensitivities is crucial for improving HCT outcomes.
SETD4, a novel lysine methyltransferase (Wang et al., NAR Cancer, 2022), has been shown to play a critical role in radiation response. Previous studies demonstrated that its deletion extends mouse survival after 8 Gy TBI by significantly preserving BM niche integrity while also increasing the radiosensitivity of hematopoietic cells (Feng et al., Int J Radiat Oncol Biol Phys, 2020). Additionally, Setd4 deletion has been shown to enhance donor cell competitiveness post-transplant and delay radiation-induced tumorigenesis, offering notable benefits for HCT success and secondary tumor prevention (Feng et al., DNA Repair, 2020).
Recently, we generated a novel Setd4 knock-in mouse model expressing an enzymatically inactive SETD4 variant. Our findings indicate that targeted enzymatic suppression of SETD4 produces radiosensitivity effects comparable to those observed in knockout models. These results support the potential for a clinically translatable strategy to modulate radiation responses by specifically inhibiting SETD4’s enzymatic activity.
Dr. Wang’s laboratory ongoing research aims to elucidate the molecular substrates and signaling pathways involved in mediating these differential effects on hematopoietic and niche cells. Our ultimate goal is to advance radioprotection and hematopoietic recovery strategies.
- Aim 1: To demonstrate that loss of SETD4 enzymatic function sensitizes recipient HSPCs to radiation.
- Aim 2: To evaluate the protective effects of SETD4 loss on the BM niche during radiation exposure.
- Aim 3: To identify the molecular substrates and signaling pathways mediating these differential effects on hematopoietic and niche cells.
This research has the potential to transform treatments for blood cancers and hematopoietic disorders, prevent BM failure caused by acute radiation exposure, and revolutionize approaches in radioprotection and regenerative medicine.