Our Research
Pancreatic ductal adenocarcinoma (PDAC), the most common type of pancreatic malignancies, is among the deadliest of all epithelial cancers. Like many other solid cancer types, PDAC has extensive genomic heterogeneity. Genomic studies uncovered important cancer-initiating oncogenes and tumor suppressor genes. However, genetic alterations alone cannot fully explain the heterogeneous nature of drug resistance and metastasis in PDAC. In this light, mutations that can be leveraged to guide therapy for most patients don’t exist. Beyond the genetic alterations, malignant cell state defined by RNA-Seq has in recent years emerged as a clinically relevant feature in PDAC. Given the complexity of the tumor microenvironment, drivers of malignant cell states most likely extend beyond cell intrinsic genomic alterations.
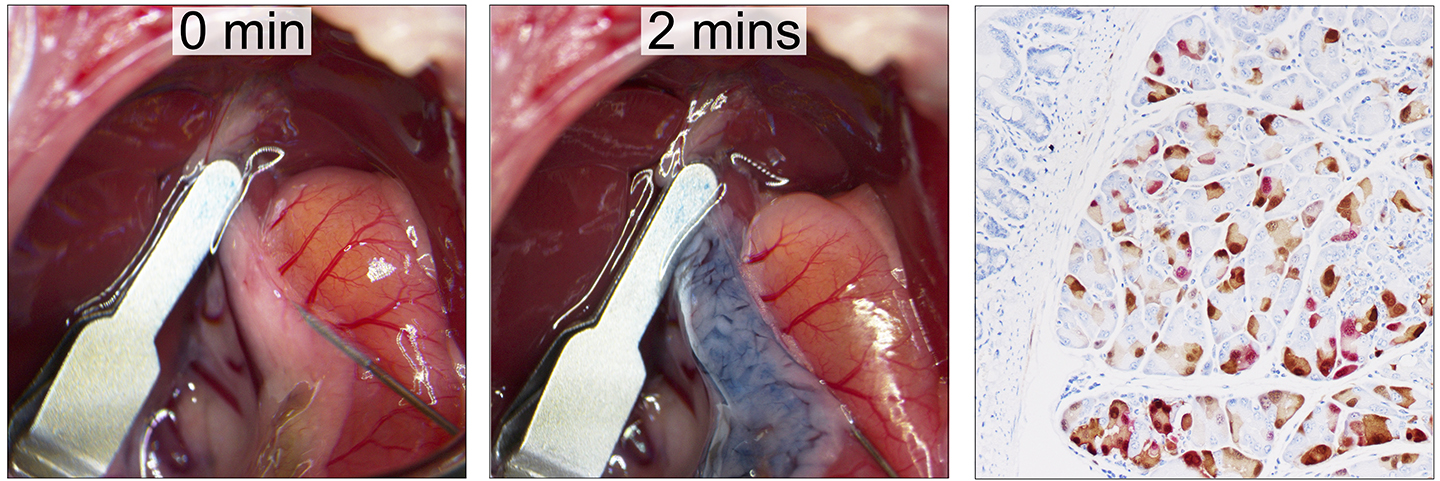
The goal of our lab is to understand how PDAC progresses within the unique tumor microenvironment it creates. We are particularly interested in understanding the mechanism by which cancer cell extrinsic factors of the tumor microenvironment drive the aggressive cancer cell phenotype and differentiation of disease subtypes that are resistant to treatment. To enable a robust and unbiased interrogation of the gene function in malignant cells, we implement methods that integrate genetically engineered mouse models of PDAC, surgical delivery of engineered viral vectors into the murine pancreas, CRISPR/Cas9-based somatic genome editing, and quantitative genomics through deep sequencing. The unique approach allows us to induce autochthonous PDAC from somatic pancreatic epithelium with engineered Kras variants through homology directed repair while disrupting an investigated gene-of-interest simultaneously. Using this strategy, our recent findings show that hypoxia can promote the aggressive basal-like PDAC subtype through the suppression of histone demethylases. In addition to hypoxia, we are also interested in exploring other cancer cell extrinsic factors that play important roles in PDAC subtype determination and disease progression.
Images: Pancreatic ductal injection infusing trypan blue through a 30-gauge needle at 0 and 30 second (left and middle). Pancreatic section of a ROSA26-Motley mouse receiving viral-cre. Immunohistochemistry of YFP and Tomato shows a mosaic pattern of expression 3 days post-surgery.